Lammps simulations using polynomial machine learning potentials¶
lammps-polymlp package is required to use polynomial machine learning potentials in lammps code. lammps-polymlp package for LAMMPS software is available on GitHub [lammps-polymlp-package]. Machine learning potentials for a wide range of systems can be found in Machine Learning Potential Repository. If you use lammps-polymlp package and machine learning potentials in the repository for academic purposes, please cite the following article [1].
[1] A. Seko, “Systematic development of polynomial machine learning potentials for elemental and alloy systems”, J. Appl. Phys. 133, 011101 (2023).
Building lammps with lammps-polymlp package¶
Unzip lammps-polymlp.tar.gz, and then copy all the components in the lammps-polymlp package to the latest lammps source code directory as
> cp -r lammps-polymlp-package/lib/polymlp $(lammps_src)/lib > cp -r lammps-polymlp-package/src/POLYMLP $(lammps_src)/src
Add “polymlp” to variable PACKAGE defined in $(lammps_src)/src/Makefile and activate polymlp package as
> cat $(lammps_src)/src/Makefile PACKAGE = \ adios \ amoeba \ ... poems \ polymlp \ ptm \ ... ml-iap \ phonon ... > ulimit -s unlimited > cd $(lammps_src)/src > make yes-polymlpBuild lammps binary files (It requires approximately ten minutes to one hour for compiling polymlp_gtinv_data.cpp.)
> make serial -j 36
(lammps-polymlp-package is tested using LAMMPS_VERSION “lammps-23Jun2022”)
Lammps input commands to specify a machine learning potential¶
The following lammps input commands specify a machine learning potential.
pair_style polymlp
pair_coeff * * mlp.lammps Ti Al
Scaling with respect to number of atoms¶
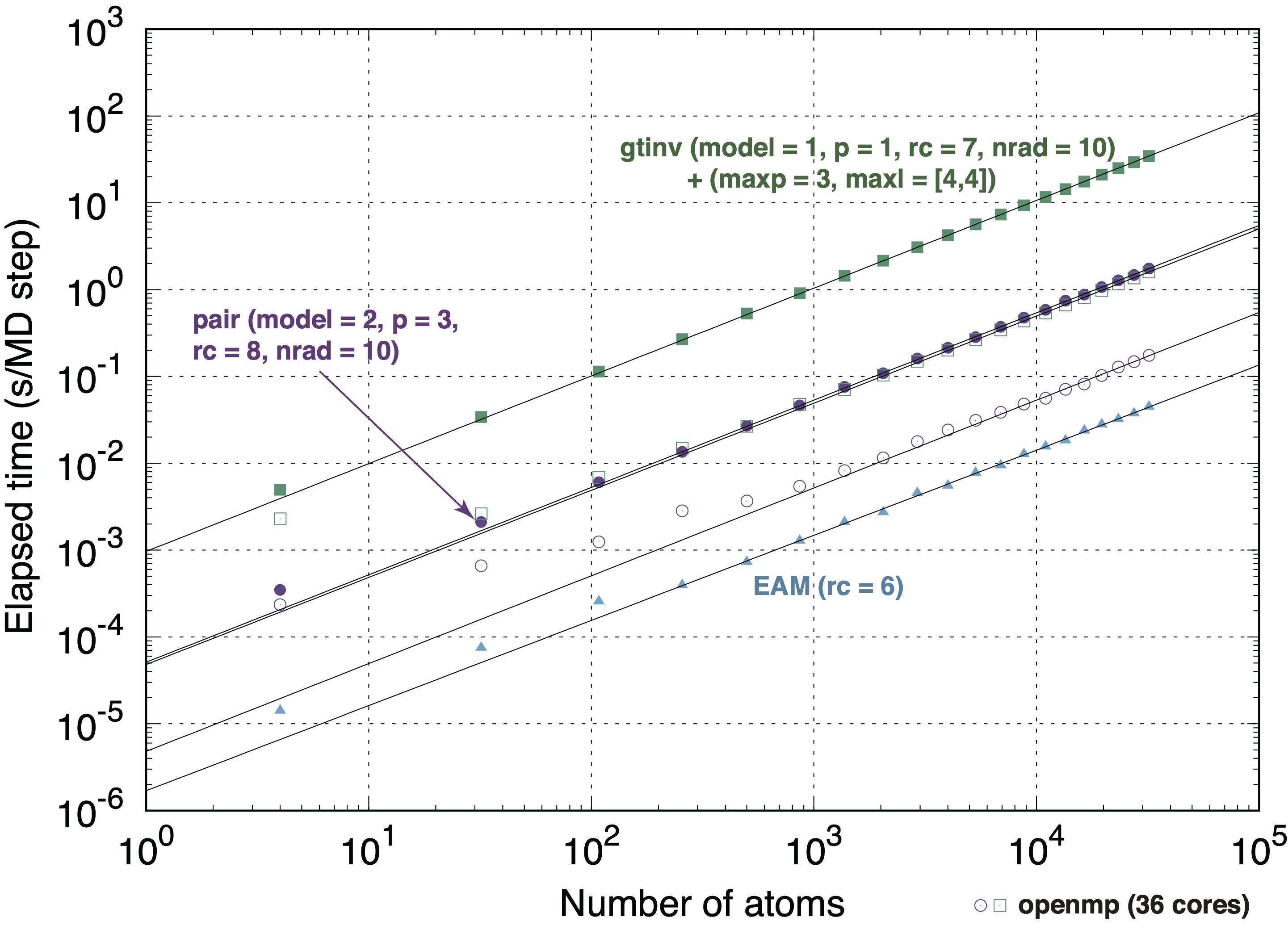
Linear scaling with respect to the number of atoms is achieved in MLPs with pairwise structural features and those with group theoretical invariants derived from spherical harnomics. So the required computational time can be estimated by t * n_atoms * n_steps, where t denotes the required computational time for single step and single atom.
Parameters controlling accuracy and computational cost of machine learning potentials¶
Input parameters used to construct a machine learning potential can be found in the correponding potential file mlp.lammps. Input parameters that control the accuracy and the computational cost are listed as follows.
1. Structural features representing neighboring atomic distribution
Cutoff radius (Angstrom)
Structural feature type (pairwise features or group theoretical invariants derived from spherical harmonics)
Radial function type (Gaussian or spherical Bessel functions)
2. Energy model with respect to structural features
Polynomial model type (1: only powers of features, 2: cross terms of all features, 3: cross terms of pairwise features)
Polynomial order of energy model
3. Truncation of group theoretical invariants
Maximum angular number of spherical harmonics
Maximum order of invariants
Whether or not symmetric invariants are used
A more detailed description can be found in the article [1].
[1] A. Seko, A. Togo and I. Tanaka, “Group-theoretical high-order rotational invariants for structural representations: Application to linearized machine learning interatomic potential”, Phys. Rev. B 99, 214108 (2019).